Czym jest fenyloketonuria? Kompendium wiedzy o chorobie

Fenyloketonuria (PKU) to ciągle najczęściej spotykana wrodzona wada metabolizmu aminokwasów. Z powodu braku lub znacznego niedoboru enzymu hydroksylazy fenyloalaninowej (PAH), we krwi gromadzą się wysokie stężenia fenyloalaniny (Phe), które nieleczone prowadzą do nieodwracalnego uszkodzenia układu nerwowego.
Co to jest fenyloketonuria?
Fenyloketonuria (PKU) to wrodzona wada metabolizmu aminokwasu o nazwie fenyloalanina (Phe). Fenyloalanina należy do grupy aminokwasów egzogennych, co oznacza, że organizm nie potrafi jej sam wytworzyć i musi ona zostać dostarczona wraz z pożywieniem (białkiem). Jest ona niezbędna do budowy białek ustrojowych, jednak jej nadmiar musi zostać przekształcony.
W zdrowym organizmie za ten proces odpowiada enzym o nazwie hydroksylaza fenyloalaninowa (PAH), który przekształca fenyloalaninę w inny aminokwas – tyrozynę. Tyrozyna jest kluczowa dla produkcji ważnych neuroprzekaźników (dopaminy, noradrenaliny) oraz melaniny (barwnika skóry i włosów). U osób z PKU proces ten jest zaburzony z powodu mutacji w genie PAH. W efekcie fenyloalanina gromadzi się we krwi i tkankach, stając się toksyczna dla rozwijającego się mózgu i całego ośrodkowego układu nerwowego (OUN).
Kluczowe fakty:
- Fenyloketonuria wykrywana jest w Polsce u każdego noworodka dzięki testom z tzw. suchej kropli krwi.
- PKU jest chorobą nieuleczalną, ale przy jej wczesnym rozpoznaniu i odpowiedniej diecie można zapobiec wystąpieniu objawów i ciężkich powikłań.
Sprawdź także: Objawy fenyloketonurii i diagnostyka – jak rozpoznać PKU?
Dlaczego chorujemy? Dziedziczenie fenyloketonurii
Za rozwój fenyloketonurii odpowiada predyspozycja genetyczna. Choroba ta jest dziedziczona w sposób autosomalny recesywny, co w praktyce oznacza, że za jej rozwój odpowiedzialna jest mutacja genu przekazana dziecku przez oboje rodziców. Przyczyną choroby są mutacje w genie kodującym enzym – hydroksylazę fenyloalaninową – który bierze udział w metabolizmie aminokwasu fenyloalaniny, pochodzącego z diety i katalizuje jej konwersję do tyrozyny.
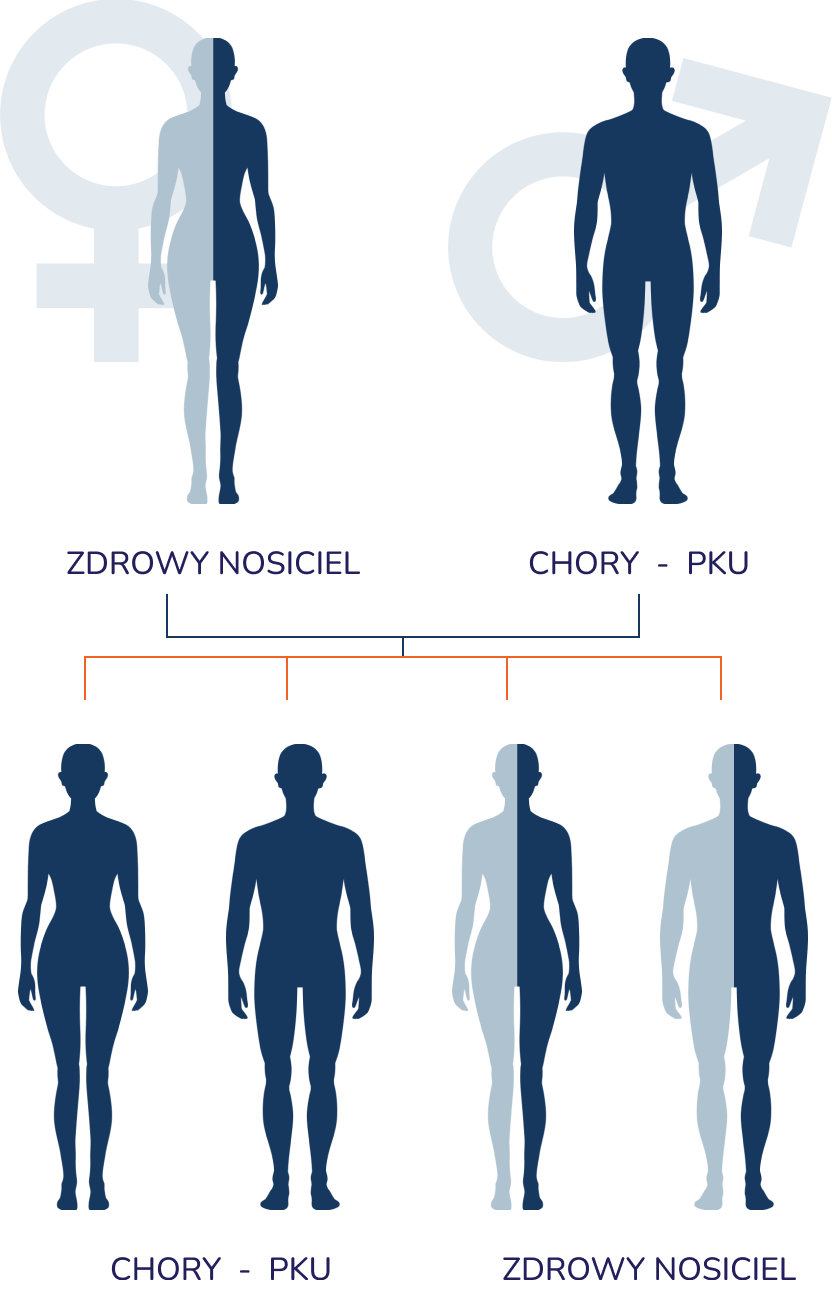
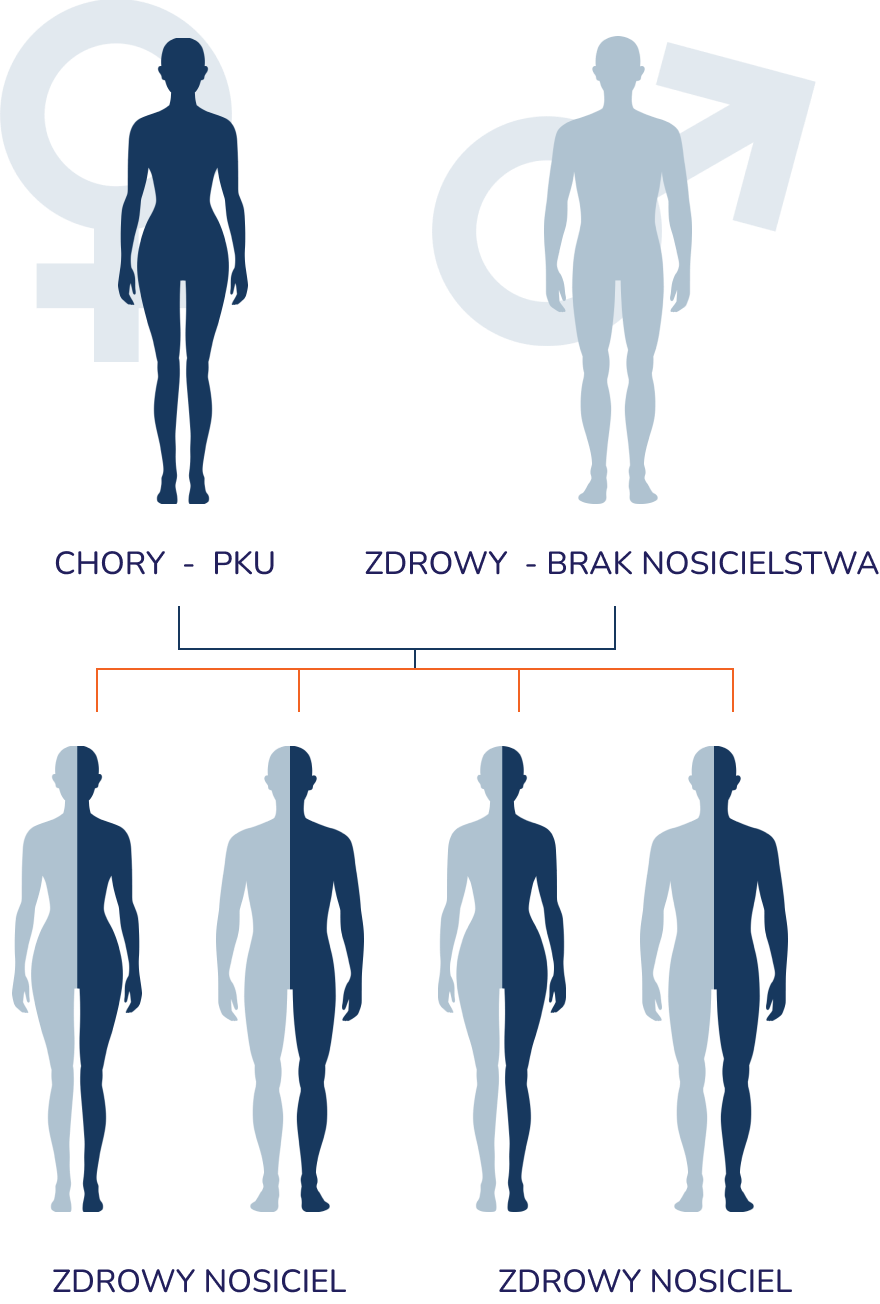
Mechanizm dziedziczenia fenyloketonurii w praktyce
- Dziedziczenie autosomalne – mutacja związana jest z chromosomami innymi niż chromosomy płci (autosomami).
- Dziedziczenie recesywne – aby choroba wystąpiła, dziecko musi otrzymać dwie kopie uszkodzonego genu (jedną od matki i jedną od ojca).
- Nosicielstwo (heterozygoty) – rodzice zazwyczaj są zdrowymi nosicielami (heterozygotami) i posiadają mutację tylko na jednym chromosomie, dlatego sami nie wykazują objawów choroby i często nie są świadomi jej obecności w swoim kodzie genetycznym. Jeśli dwie takie osoby (nosiciele) zostaną rodzicami i przekażą dziecku chromosom z mutacją, dochodzi do rozwoju fenyloketonurii.
Jak często występuje fenyloketonuria?
Częstość występowania fenyloketonurii w Polsce, podobnie jak w innych krajach Europy Środkowej, wynosi od 1:7000 do 1:8000. Jest to najczęściej występująca choroba wśród wrodzonych wad metabolizmu. Częstość występowania łagodnych postaci hiperfenyloalaninemii w populacji polskiej jest około 5 razy mniejsza w porównaniu z postacią klasyczną.
Sprawdź także: Leczenie fenyloketonurii na świecie – nowoczesne terapie i leki
Rodzaje fenyloketonurii
W zależności od stężenia fenyloalaniny we krwi oraz stopnia uszkodzenia enzymu PAH wyróżniamy kilka postaci choroby:
Fenyloketonuria klasyczna
To najcięższa postać choroby, w której aktywność enzymu hydroksylazy fenyloalaninowej w bioptatach komórek wątroby jest niższa niż 1%. W konsekwencji stężenie fenyloalaniny we krwi jest znacznie wyższe – na zwykłej diecie sięga powyżej poziomu >1200 µmol/l. Pacjenci w tej grupie są niezwykle wrażliwi na wszelkie odstępstwa bądź zaniedbania terapii. Tak wysoki poziom fenyloalaniny przyczynia się do licznych zaburzeń neurologicznych, takich jak zaburzenia mielinizacji i uszkodzenia istoty białej układu nerwowego. U nieleczonych niemowląt zauważalne są także zaburzenia w istocie szarej, przede wszystkim w korze ruchowej, przedruchowej, wzgórzu i hipokampie.
Łagodna fenyloketonuria
Należy do nieco łagodniejszych postaci choroby. Aktywność enzymu hydroksylazy fenyloalaninowej wynosi od 1 do 3%, dzięki czemu stężenie fenyloalaniny we krwi u tych niemowląt na zwykłej diecie jest na poziomie 600–1200 µmol/l. Pacjenci z łagodną fenyloketonurią wykazują się nieco wyższą tolerancją na wysokie stężenia fenyloalaniny we krwi, jednak nadal do prawidłowego rozwoju niezbędna jest odpowiednia terapia i unikanie nadmiaru fenyloalaniny w diecie.
Łagodna hiperfenyloalaninemia (HPA)
Zdecydowanie najłagodniejszy typ fenyloketonurii. W przypadku HPA aktywność enzymu odpowiedzialnego za metabolizm fenyloalaniny sięga nawet od 3 do 6%. W przypadku pacjentów z łagodną hiperfenyloalaninemią stężenie fenyloalaniny we krwi na zwykłej diecie zazwyczaj nie przekracza 600 µmol/l. Jednakże nadal, do prawidłowego rozwoju dzieci, ważne jest przestrzeganie odpowiednio dobranej terapii i utrzymywanie poziomu fenyloalaniny we krwi na ustalonych wartościach.
Fenyloketonuria matczyna
Nieco innym rodzajem fenyloketonurii, na jaką narażone są niemowlęta, jest fenyloketonuria matczyna. Występuje ona, gdy na fenyloketonurię choruje kobieta ciężarna i nie przestrzega odpowiedniej diety eliminacyjnej. W tej sytuacji również występuje brak aktywności enzymu hydroksylazy fenyloalaninowej. Jeśli mama nie przestrzega terapii, dochodzi do podniesienia poziomu fenyloalaniny we krwi, co może przyczynić się do uszkodzenia płodu. U takich niemowląt najczęściej obserwuje się przede wszystkim opóźnienie rozwoju układu nerwowego, zaburzenia rozwoju umysłowego i motorycznego, a także wady serca.
| Typ fenyloketonurii | Poziom Phe we krwi (mg/dl) | Charakterystyka |
|---|---|---|
| PKU Klasyczna | > 20 mg/dl | Całkowity brak lub minimalna aktywność enzymu. Wymaga rygorystycznej diety. |
| PKU Łagodna | 10–20 mg/dl | Częściowa aktywność enzymu, nieco większa tolerancja na Phe w diecie. |
| Łagodna Hiperfenyloalaninemia (HPA) | 2–10 mg/dl | Najlżejsza postać, często wymaga jedynie monitorowania poziomu Phe bez ścisłego leczenia. |
Podstawy leczenia fenyloketonurii – na czym polega dieta PKU?
Podstawą leczenia PKU jest dieta niskofenyloalaninowa, która ogranicza negatywny wpływ aminokwasu na układ nerwowy. Odpowiednio skomponowany jadłospis umożliwia dzieciom prawidłowy wzrost i rozwój, a także funkcjonowanie zbliżone do normalnego. Jest to szczególnie ważne w przypadku niemowląt i małych dzieci, u których to układ nerwowy dopiero się kształtuje.
Ten model żywienia obowiązuje chorych przez całe życie. Podaż fenyloalaniny w codziennej diecie jest ustalana dla każdego pacjenta indywidualnie i zmienia się wraz z wiekiem.
Sprawdź także: 6 pomysłów, jak uniknąć dietetycznej monotonii w diecie PKU
Kluczowe filary diety niskofenyloalaninowej:
- Preparaty PKU – specjalistyczne mieszanki aminokwasowe (bez Phe) wzbogacone o witaminy i minerały. Zastępują one białko naturalne i muszą być spożywane codziennie w dawkach ustalonych przez specjalistę.
- Indywidualizacja – podaż fenyloalaniny jest dobierana dla każdego pacjenta indywidualnie – zbyt wysoka jest toksyczna, ale zbyt niska może zaburzać m.in. pracę tarczycy.
- Żywność niskobiałkowa – zastępowanie tradycyjnych produktów (mięsa, nabiału, pieczywa) ich specjalistycznymi odpowiednikami PKU o obniżonej zawartości białka.
Lista produktów dozwolonych i zabronionych na diecie niskofenyloalaninowej
| PODZIAŁ PRODUKTÓW SPOŻYWCZYCH W DIECIE NISKOFENYLOALANINOWEJ | |
|---|---|
| DOZWOLONE | ZABRONIONE |
|
|
Sprawdź także: Dieta przy fenyloketonurii: produkty zakazane i dietetyczne grzechy
Fenyloketonuria, choć jest chorobą nieuleczalną i wymagającą ogromnej dyscypliny, nie musi oznaczać ograniczeń w realizacji życiowych planów. Kluczowym czynnikiem sukcesu jest wczesne rozpoznanie w ramach badań przesiewowych oraz natychmiastowe wdrożenie nowoczesnej dietoterapii. Dzięki ścisłej współpracy z lekarzami, regularnemu stosowaniu preparatów PKU oraz kontroli stężenia fenyloalaniny, osoby z PKU mogą cieszyć się pełnią zdrowia, rozwijać swoje pasje i funkcjonować w społeczeństwie na równi z rówieśnikami. Współczesny rynek żywności niskobiałkowej stale się rozwija, ułatwiając pacjentom prowadzenie urozmaiconej i smacznej diety każdego dnia.
Najważniejsze pytania o PKU
Czy PKU mija z wiekiem?
Nie, fenyloketonuria towarzyszy pacjentowi przez całe życie. Choć dawniej uważano, że dietę można poluzować po zakończeniu rozwoju mózgu, współczesna wiedza medyczna zaleca model „Diet for life”. Utrzymywanie niskiego stężenia fenyloalaniny u dorosłych jest niezbędne, aby uniknąć problemów z koncentracją, obniżonego nastroju czy innych zaburzeń neurologicznych.
Czy osoby z PKU mogą uprawiać sport?
Tak – aktywność fizyczna jest wręcz wskazana dla ogólnego stanu zdrowia. Sportowcy z PKU muszą jedynie pamiętać o odpowiednim zwiększeniu podaży energii i preparatów aminokwasowych, aby zapobiec procesom katabolicznym (rozpadowi białek własnych), które mogłyby podnieść poziom Phe we krwi.
Czy kobieta z fenyloketonurią może urodzić zdrowe dziecko?
Kobieta z PKU może urodzić zdrowe dziecko pod warunkiem zachowania rygorystycznej diety i utrzymania stężenia fenyloalaniny poniżej 6 mg% (optymalnie 2–4 mg%). Niezbędne jest planowanie ciąży z co najmniej trzymiesięcznym wyprzedzeniem, aby od początku zapewnić płodowi optymalne warunki rozwoju. Odpowiednia terapia pozwala na urodzenie dziecka, które pod względem fizycznym i intelektualnym nie różni się od rówieśników.
Czy dziecko z fenyloketonurią może być karmione piersią?
Tak, karmienie piersią jest możliwe i zalecane, ale musi odbywać się pod ścisłą kontrolą stężenia Phe we krwi noworodka. Mleko matki zawiera fenyloalaninę, dlatego zazwyczaj łączy się karmienie naturalne z podawaniem preparatu bezfenyloalaninowego w odpowiednich proporcjach ustalonych przez lekarza.
Bibliografia:
- Choroby i wady wykrywane w badaniach przesiewowych, Instytut Matki i Dziecka, Zakład Badań przesiewowych, http://przesiew.imid.med.pl/fenyloketonuria.html
- Fenyloketonuria – diagnostyka, leczenie, wybrane problemy kliniczne, lek. Bożena Didycz, dr hab. n. med. Mirosław Bik-Multanowski, prof. UJ, Pediatria po Dyplomie, 2017, 02.
- Centrum Badań Genetycznych, www.genesis.pl Narodowe Centrum Edukacji Żywieniowej, Instytut Żywności i Żywienia, https://ncez.pl/dzieci-i-mlodziez/dzieci-0-3/fenyloketonuria-----rzadka-choc-czesta-wada-metabolizmu
- Podstawy naukowe żywienia w szpitalach. M. Jarosz i wsp. Instytut Żywności i Żywienia, 2001.
- Żywienie człowieka zdrowego i chorego cz. 2. M. Grzmisławski i wsp. Wydawnictwo Naukowe PWN 2012.