Historia leczenia fenyloketonurii – kartki z kalendarza
- Data publikacji: 22.02.2019
- 20 min
Jak odkryto PKU?
Historia leczenia fenyloketonurii sięga lat 40-tych XX wieku. Choroba ta została po raz pierwszy opisana w 1934 r. przez norweskiego lekarza i biochemika Ivara Asbjørna Føllinga na podstawie tygodni obserwacji dwójki rodzeństwa z głęboką niepełnosprawnością intelektualną.
Dzieci zwracały uwagę blond włosami, jasnoniebieskimi tęczówkami i jasną karnacją skóry, o których dzisiaj wiadomo, że są charakterystyczne dla nieleczonej fenyloketonurii (tzw. zjawisko „rozcieńczenia barwnika”) oraz nieprzyjemnym, określanym jako mysi, zapachem. Fölling zaobserwował, że mocz rodzeństwa po dodaniu chlorku żelaza przejściowo zabarwiał się na zielono (czego nie obserwowano wcześniej u ludzi zdrowych).
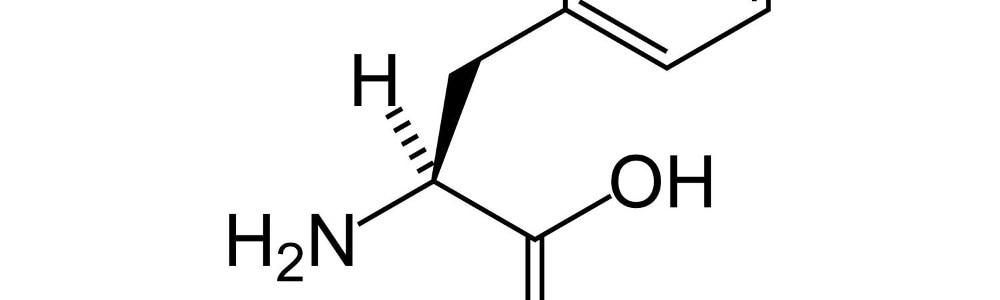
Za zmianę barwy moczu i swoisty zapach odpowiedzialny okazał się kwas o–hydroksyfenylooctowy, będący produktem ubocznym metabolizmu aminokwasu fenyloalaniny. Pięć miesięcy od pierwszej wizyty matki z chorymi dziećmi, Fölling dla nazwania opisanej przypadłości użył określenia imbecillitas phenylpyruvica, co było znaczącym momentem dla całej historii leczenia fenyloketonurii i badań nad tą chorobą.
W 1937 roku George Jervis, amerykański lekarz i badacz zastosował nową nazwę choroby - phenylpyrouvicoligophrenia. W tym samym roku Lionel Penrose, angielski genetyk, zidentyfikował autosomalny recesywny sposób jej dziedziczenia i zasugerował skrócenie nomenklatury do phenylketonuria (w skrócie PKU), która funkcjonuje do dnia dzisiejszego.
Historia leczenia fenyloketonurii: w poszukiwaniu winowajcy
Odkrycia Föllinga zachęciły do dalszych badań nad chorobą. W 1953 r. George Jervis zidentyfikował wrodzony defekt enzymatyczny w wątrobie chorych na PKU, który uniemożliwia przemianę fenyloalaniny do tyrozyny. Za blokowanie tej przemiany odpowiedzialny okazał się brak aktywności enzymu hydroksylazy fenyloalaninowej powodujący gromadzenie nadmiaru fenyloalaniny we krwi.
Historia leczenia fenyloketonurii: pierwsze zastosowanie specjalistycznej diety w leczeniu PKU
Kolejnym pionierem na polu fenyloketonurii był profesor Horst Bickel, niemiecki lekarz, który zaproponował leczenie dietą niskofenyloalaninową. Był to zupełnie nowy rozdział w historii leczenia fenyloketonurii. Zastosowane kuracji żywieniowej dało spektakularne efekty terapeutyczne i miało miejsce w 1951 roku. Zespół Bickla napotkał na spore kłopoty z upowszechnieniem swoich obserwacji i artykuł dotyczący leczenia fenyloketonurii dietą ukazał się w druku dopiero w 1954 r.
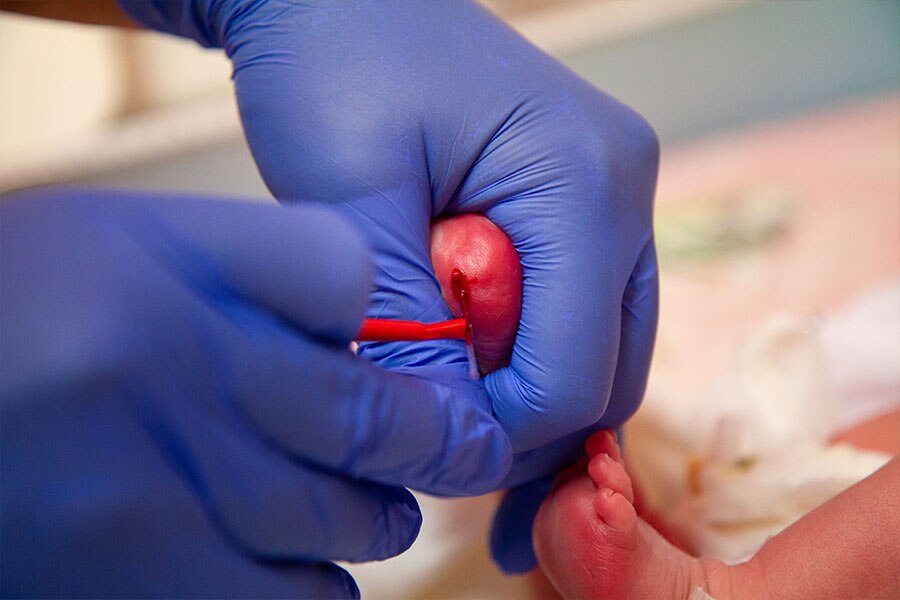
Kamień milowy w historii leczenia PKU – identyfikacja chorych noworodków
Przełomowym momentem w wykrywaniu chorych na fenyloketonurię było upowszechnienie pierwszego diagnostycznego testu przesiewowego u noworodków. W 1957 r. lekarz Willard Centerwall z Los Angeles rozpoczął stosowanie testu mokrej pieluszki (ang. wet diaper test) poprzez dodawanie do niej chlorku żelaza. Wyeliminowano w ten sposób zbieranie próbek moczu i wysyłanie ich do laboratorium. Istotną wadą testu była możliwość jego wykonania nie wcześniej niż w 5-6 tygodniu życia dziecka.
Wprowadzenie preparatu do leczenia
W roku 1958 stworzono pierwszą niskofenyloalaninową mieszankę stosowaną w leczeniu fenyloketonurii. Firma Mead Johnson wyprodukowała hydrolizat białek mleka krowiego o niskiej zawartości fenyloalaniny. Lofenalac był pierwszym preparatem przeznaczonym dla pacjentów chorych na PKU (z noworodkami włącznie) zastępującym wysokobiałkowe produkty spożywcze. Zawierał węglowodany, tłuszcze, witaminy, makro i mikroelementy oraz niewielkie ilości fenyloalaniny.
Kolejny krok w historii leczenia fenyloketonurii: uproszczenie diagnostyki poprzez zastosowanie testu bibułowego
Najważniejszym krokiem w historii leczenia fenyloketonurii w kierunku upowszechnienia diagnostyki choroby było opracowanie przez Roberta Guthriego w 1960 r. prostego testu, polegającego na pobraniu niewielkiej ilości krwi na specjalną bibułę. Dzięki niemu wykrycie podwyższonych stężeń fenyloalaniny we krwi niemowlęcia stało się możliwe wkrótce po narodzinach. Test bibułowy pozwolił identyfikować dzieci chore na PKU przed wystąpieniem szkodliwych objawów i szybkie włączenie postępowania żywieniowego.
W latach 1964 - 1982 sto siedemdziesiąt dzieci wzięło udział w programie Collaborative Study of Children with PKU prowadzonym przez Richarda Kocha z Los Angeles. Program dowiódł, że wprowadzenie postępowania dietetycznego w ciągu 14 pierwszych dni życia dziecka, skutkujące obniżeniem stężeń fenyloalaniny do wartości bezpiecznych oraz ich utrzymanie i monitorowanie, daje chorym na PKU takie same możliwości rozwoju jak zdrowym rówieśnikom.
W 1983 roku Savio Woo, badacz zajmujący się inżynierią biomedyczną, wyizolował gen hydroksylazy fenyloalaninowej.
Historia leczenia fenyloketonurii w Polsce
Instytut Matki i Dziecka w Warszawie został powołany do życia w roku 1948 r. na wniosek Ministra Zdrowia. Głównym celem utworzenia Instytutu była potrzeba oparcia organizacji ochrony zdrowia matki i dziecka w Polsce na podstawach naukowych.
Organizacja i główne kierunki działania Instytutu Matki i Dziecka (IMiD) zmieniały się od tego czasu w zależności od stanu zdrowia społeczeństwa i najpilniejszych potrzeb. Jednym z wydarzeń, mających istotne znaczenie w historii leczenia fenyloketonurii w Polsce, było rozpoczęcie w 1965 r. przez profesor Barbarę Cabalską i doktor Ninę Duczyńską badań przesiewowych w kierunku fenyloketonurii poprzez zastosowanie testu Guthriego.
Badanie pozwoliło rozpoznawać chorobę we wczesnym wieku dziecka, wdrożyć terapię żywieniową zapobiegając tym samym uszkodzeniu ośrodkowego układu nerwowego prowadzącego do niepełnosprawności intelektualnej. Przez wiele lat badanie nie było obligatoryjne. Dopiero w 1994 r. wszystkie noworodki zostały objęte obowiązkowym badaniem przesiewowym. W 1998 r. test Guthriego został zastąpiony badaniem metodą tandemowej spektrometrii mas (tandem MS/MS). Obok PKU umożliwiło to wykrywanie wielu innych wad metabolizmu.
W chwili obecnej na terenie Polski znajduje się siedem ośrodków wykonujących badania przesiewowe noworodków, co umożliwia szybkie diagnozowanie dzieci nawet z najdalszych zakątków kraju. Z roku na rok zwiększa się ilość chorób metabolicznych objętych badaniem przesiewowym.



