Fenyloalanina w nietypowych produktach, jak uniknąć nieświadomego narażenia się na zaburzenia metabolizmu?

- Data publikacji: 12.06.2019
- 30 min
Obowiązkowe badania przesiewowe, w pierwszych dniach po urodzeniu, wykrywają wrodzoną chorobę metaboliczną jaką jest fenyloketonuria. Jedynym sposobem leczenia tej choroby jest restrykcyjna dieta, w której ustala się indywidualne zapotrzebowanie dobowe na fenyloalaninę (Phe) – aminokwas spożywany z jedzeniem. Osoby chore, a w młodszym wieku ich rodzice, aby nieświadomie nie narażać się na poważne konsekwencje zaburzonego metabolizmu fenyloalaniny powinni prześledzić dostępne tabele składu wartości odżywczych produktów i zawartą w nich ilość Phe w jedzeniu.
Dlaczego tak ważne jest unikanie fenyloalaniny przy PKU?
Fenyloketonuria (w skrócie PKU) jest przekazywana dziecku przez geny pochodzące od obu rodziców. Jest to choroba spowodowana wrodzonym niedoborem lub brakiem enzymu o nazwie hydroksylaza fenyloalaninowa (PAH) odpowiedzialnego za metabolizm fenyloalaniny do innego aminokwasu - tyrozyny.
O tym, czym są enzymy i jaką rolę pełnią przeczytasz więcej tutaj.
Czym jest fenyloalanina (Phe)?
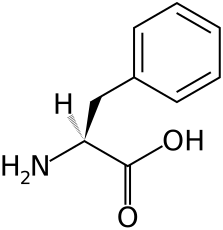
Fenyloalanina jest jednym z 20 aminokwasów kodowanych przez DNA zwierząt i roślin, które stanowią podstawowy budulec większości naturalnie występujących białek. U osób zdrowych ¾ fenyloalaniny przekształca się w inny aminokwas - tyrozynę, a jej pozostała część włączana jest do budowy innych białek potrzebnych w organizmie. U chorych na PKU fenyloalanina osiąga nawet 20-krotnie większe stężenie z powodu niemożności prawidłowego metabolizmu tego aminokwasu. W dalszej perspektywie prowadzi to do uszkodzeń ośrodkowego układu nerwowego i upośledzeń umysłowych. Jedynym sposobem postępowania w tej wrodzonej chorobie metabolicznej jest zastosowanie odpowiedniej diety o niskiej zawartości białka i kontrolowanej, minimalnej dla zdrowia ilości fenyloalaniny. Nie możemy całkowicie pozbawić organizmu fenyloalaniny, ale w przypadku tej wrodzonej choroby metabolicznej zachodzi konieczność ścisłej kontroli jej spożycia.
Fenyloalanina: normy we krwi
Okazuje się, że optymalne stężenie fenyloalaniny jest kwestią dyskusyjną i zależy od ośrodka leczniczego. Niewątpliwie kluczowy jest poziom fenyloalaniny we krwi niemowląt i małych dzieci. Zgodnie z wytycznymi brytyjskimi w tej grupie związek ten powinien być utrzymywany na poziomie od 2 do 6 mg/dl. U starszych dzieci optymalne stężenie fenyloalaniny we krwi nie jest ustalone. Z kolei eksperci z Niemiec (Niemiecka Grupa Robocza ds. Chorób Metabolicznych) uważają, że należy utrzymywać poziom fenyloalaniny we krwi w następujących zakresach:
- 0,7–4 mg/dl do 10. roku życia,
- 0,7–15 mg/dl 15. roku życia,
- 0,7–20 mg/dl po ukończeniu 15. roku życia.
Z kolej w USA zaleca się utrzymywanie fenyloalaniny na poziomie 2 – 6 mg/dl u pacjentów do 12 roku życia i następnie od 2 do 10 mg/dl po ukończeniu 12. roku życia. Jak podkreślają Eksperci, ważna jest ciągła obserwacja dziecka i przeprowadzanie regularnych badań. Wszelkie odchylenia od normy powinny być korygowane odpowiednią terapią.
Podsumowując, najważniejszy moment to okres do 12. roku życia, kiedy to zachodzi intensywny wzrost i rozwój, a także kształtowanie się układu nerwowego. W tym wieku stężenie fenyloalaniny powinno być utrzymywane w przedziale od 2 do 6 mg/dl. W przypadku starszych dzieci normy nie są już tak restrykcyjne, natomiast nie należy zapominać, że nadal fenyloalanina wpływa na czynności poznawcze. Z tego względu u starszych dzieci, młodzieży i dorosłych zaleca się utrzymywanie stężenia fenyloalaniny w zakresie od 2 do 10 - 15 mg/dl.
Czytaj więcej: Jakie powinno być stężenie fenyloalaniny u leczonego pacjenta z fenyloketonurią?
|
Zalety i wady fenyloalaniny |
|
|---|---|
|
Zalety |
Wady |
|
Pomoc w depresji |
Niewskazana dla osób z PKU |
|
Redukcja masy działa |
Może powodować nudności |
|
Redukcja bólu |
|
|
Pomoc przy uzależnieniu od substancji |
|
Jak uniknąć konsekwencji zdrowotnych zaburzeń metabolizmu Phe?
U dziecka pozytywnie zdiagnozowanego w badaniach przesiewowych w pierwszych dniach po urodzeniu należy niezwłocznie wprowadzić dietę ubogą w fenyloalaninę. Musi ona zawierać jej dokładnie tyle, ile organizm potrzebuje do wzrostu i przemian białkowych w organizmie, a jednocześnie, aby uniknąć niedorozwoju umysłowego wywołanego nadmiarem toksycznych substancji, powstałych ze źle metabolizowanej fenyloalaniny, nieodwracalnie uszkadzających tkankę mózgu.
Aby mieć świadomość, jakie ilości dobowego spożycia fenyloalaniny są dobrze tolerowane u osoby chorej na PKU, stosuje się przelicznik w zależności od stopnia niedoboru enzymu PAH, od wieku i masy ciała, co obrazuje poniższa tabela podająca przelicznik dla dziecka w wieku 5 lat. Lekarz prowadzący i dietetyk specjalizujący się w chorobach metabolicznych określają to, w jakiej ilości fenyloalanina jest tolerowana przez konkretny organizm. Do bilansowania restrykcyjnej diety PKU stosuje się specjalistyczną żywność PKU dostosowaną do wieku i zapotrzebowania organizmu na białko i inne ważne składniki odżywcze.
W zależności od tego czy w organizmie występuje brak czy jest za mało enzymu PAH, rozróżniamy różne typy choroby, w uproszczeniu:
|
Postać choroby |
Stężenie fenyloalaniny w surowicy krwi przed leczeniem (μmol) |
Tolerancja fenyloalaniny w diecie w wieku 5 lat (mg/kg masy ciała/dobę) |
|
Klasyczna fenyloketonuria |
> 1200 |
10-20 |
|
Łagodna fenyloketonuria |
600 - 1200 |
20 - 50 |
|
Łagodna hiperfenyloalaminemia |
600 |
50 - 100 |
Ważną zasadą, o której należy pamiętać jest to, aby dopuszczoną w pożywieniu ilość Phe rozdzielić równomiernie na 3–5 posiłków. To ważne, ponieważ fenyloalanina spożyta w formie jednorazowej dawki może doprowadzić do zachwiania równowagi metabolicznej organizmu.
W jakich produktach występuje fenyloalanina?
Fenyloalanina jest aminokwasem, który w największej ilości znajduje się w produktach spożywczych bogatych w białko. Chodzi przede wszystkim o mięso i jego przetwory, mleko i jego przetwory oraz produkty, do których dodawane jest mleko w proszku, ryby, całe jaja, rośliny strączkowe, czyli fasola, groch, soja, soczewica oraz nasiona i orzechy. Również produkty zbożowe wbrew pozorom zawierają zbyt duże ilości białka jak dla osoby chorej na tę wrodzoną chorobę metaboliczną. Tak więc zwykłe produkty zbożowe, takie jak pieczywo, mąka, kasze, płatki, makarony i pieczywo cukiernicze, również nie mogą być spożywane w diecie PKU. Ponadto osoby z fenyloketonurią nie mogą jeść zwykłej czekolady, żelatyny oraz żadnych produktów, do których dodany jest aspartam czyli E951, ze względu na to, iż w jego skład wchodzą dwa aminokwasy: fenyloalanina i kwas asparaginowy.
Więcej o zasadach diety przy fenyloketonurii przeczytasz tutaj.
Jak unikać narażenia się na nadmiar fenyloalaniny w diecie PKU w nietypowych produktach?
Należy zwracać uwagę nie tylko na unikanie podstawowych grup produktów wysokobiałkowych, ale także na te z pozoru o niskim lub zerowym stężeniu białka. Produkty, do których dodawany jest aspartam, w wielu przypadkach nie kojarzą się i nie są produktami zawierającymi białka. Dlatego należy uważnie czytać etykiety wszelkich produktów spożywczych, w tym napojów typu light, do których może być dodawany ten popularny słodzik. Patrząc na tabele składu produktów, możemy zauważyć, że różnica w zawartości fenyloalaniny w „niewinnym” szczypiorku czy czosnku w porównaniu do np. zielonego groszku nie jest aż tak bardzo znacząca w przeliczeniu na 100 g.
Tabela poniżej przedstawia zawartości fenyloalaniny w wybranych niskobiałkowych i dozwolonych produktach w diecie PKU oraz dla kontrastu kilka wysokobiałkowych.
|
Produkt/100g |
Fenyloalanina (mg) |
Energia (kcal) |
Białko (g) |
|
Dynia |
47 |
28 |
1,3 |
|
Cebula |
62 |
30 |
1,4 |
|
Burak |
81 |
38 |
1,8 |
|
Kapusta biała |
72 |
29 |
1,7 |
|
Korzeń pietruszki |
92 |
38 |
2,6 |
|
Brokuły |
124 |
27 |
3,0 |
|
Fasolka szparagowa |
123 |
29 |
3,0 |
|
Kiełki fasoli mung |
167 |
29 |
3,0 |
|
Brukselka |
185 |
37 |
4,7 |
|
Jarmuż |
190 |
29 |
3,3 |
|
Natka pietruszki |
200 |
41 |
4,4 |
|
Szczypiorek |
253 |
29 |
4,1 |
|
Czosnek |
270 |
146 |
6,4 |
|
Groszek zielony< |
330 |
75 |
6,7 |
|
Bagietki francuskie pełnobiałkowe |
434 |
283 |
8,7 |
|
Bagietka niskobiałkowa PKU |
30 |
236 |
0,8 |
|
Groch (nasiona suche) |
1172 |
293 |
23,8 |
|
Soczewica czerwona (nasiona suche) |
1380 |
327 |
25,4 |
|
Soja (nasiona suche) |
1670 |
382 |
34,3 |
Warto zajrzeć: Słodziki - rodzaje i bezpieczeństwo stosowania przy fenyloketonurii
Objawy nadmiaru fenyloalaniny w diecie
W przypadku osób zdrowych nie ma mowy o nadmiarze fenyloalaniny w diecie. Sytuacja jest jednak odmienna w przypadku chorych na fenyloketonurię (PKU). Z powodu zaburzeń w szlakach metabolicznych, u chorych z PKU, fenyloalanina spożywana w ilościach większych niż indywidualny poziom tolerancji, wpływa szkodliwie na zdrowie.
Przede wszystkim dotyczy to ośrodkowego układu nerwowego, w którym u małych dzieci i noworodków może dojść do wystąpienia nieodwracalnych uszkodzeń. Nadmiar fenyloalaniny w diecie jest związany z zaburzonym funkcjonowaniem mózgu i obniżoną sprawnością intelektualną. U chorych może występować małogłowie i nieprawidłowości w budowie twarzy (w tym osadzenia oczu i uszu oraz kształtu nosa i ust.
Pacjenci cierpią także na napady padaczkowe, zaburzenia mowy, chodu czy zachowań np. z powodu autyzmu czy agresji. Charakterystyczne jest występowanie jasnych włosów, skóry i tęczówek oka, co jest związane z zaburzonym powstawaniem barwnika - melaniny. Typowe jest również występowanie mysiego zapachu moczu i potu.
Niedobór fenyloalaniny w diecie
Warto także podkreślić, że zarówno nadmiar, jak i niedobór fenyloalaniny w diecie chorych jest szkodliwy. Każdy pacjent z PKU ma ustalone indywidualne zapotrzebowanie na fenyloalaninę. Wartość zapotrzebowania zależy od aktywności hydroksylazy fenyloalaninowej u danej osoby. Może się to wydawać zaskakujące, ale nawet w fenyloketonurii pewne ilości fenyloalaniny są niezbędne.
Fenyloalanina jest jednym z 20 aminokwasów, który bierze udział w budowaniu naturalnie występujących białek organizmu człowieka. Fenyloalanina należy do aminokwasów egzogennych, co oznacza, że musi być dostarczana wraz z pożywieniem. W organizmie człowieka jest wykorzystywana przede wszystkim do syntezy tyrozyny. Odpowiedni poziom fenyloalaniny umożliwia powstawanie tyrozyny i w konsekwencji prawidłowe funkcjonowanie przysadki mózgowej czy tarczycy. Niedobór fenyloalaniny uniemożliwia powstanie tyrozyny, powoduje niedoczynność tarczycy, odczuwanie przewlekłego zmęczenia, a także zaburzenia nastroju.
W przypadku chorych z fenyloketonurią kluczowe jest przestrzeganie odpowiedniego reżimu dietetycznego. Aby uniknąć licznych zaburzeń, należy unikać nadmiaru fenyloalaniny, ale ważne jest także spożywanie minimalnych ilości tego aminokwasu. W komponowaniu diety pomoże lekarz prowadzący i dietetyk kliniczny z odpowiednim doświadczeniem w terapii chorób metabolicznych.
Źródła:
- Zasady żywienia w PKU, https://zasadyzywienia.pl/tag/tabele-pku
- Charakterystyka Molekularna Zmian w Genach Kodujących Hydroksylazę Fenyloalaninową oraz Syntazę Tetrahydrobiopterynową w Populacji Polskiej, Instytut Matki i Dziecka, Zakład Genetyki Medycznej, T. Mazurczak i wsp.
- Fenyloketonuria - badania przesiewowe oraz leczenie
- Aktualne (2000) stanowisko ekspertów National Institutes of Health. NIH Consensus Statement Online (October 16-18, 2000); 17 (3): 1-27




