Fenyloketonuria dziś – czy daje objawy?

- Data publikacji: 21.06.2019
- 30 min
Fenyloketonuria (PKU) to ciągle najczęściej spotykana wrodzona wada metabolizmu aminokwasów. Zaburzenia dotyczą aminokwasu zwanego fenyloalaniną (Phe). Z powodu błędu metabolicznego, spowodowanego całkowitym brakiem lub znacznym ograniczeniem aktywności enzymu hydroksylazy fenyloalaninowej (PAH), we krwi gromadzą się wysokie stężenia fenyloalaniny (Phe), które działają toksycznie na ośrodkowy układ nerwowy (OUN).
Co to jest fenyloketonuria? Przyczyny choroby
PKU dziedziczona jest autosomalnie recesywnie, co oznacza, że oboje rodziców są nosicielami zmutowanego (uszkodzonego) genu. Jest chorobą nieuleczalną, ale przy jej wczesnym rozpoznaniu można zapobiec wystąpieniu objawów i ciężkich powikłań. Przyczyną choroby są mutacje w genie kodującym enzym – hydroksylazę fenyloalaninową – który bierze udział w metabolizmie aminokwasu fenyloalaniny, pochodzącego z diety i katalizuje jej konwersję do tyrozyny. W uproszczonej klasyfikacji (w zależności od stężenia fenyloalaniny we krwi) wyróżnia się 3 postacie choroby:
- klasyczną fenyloketonurię,
- łagodną fenyloketonurię,
- łagodną hiperfenyloalaninemię (HPA).
Klasyczna fenyloketonuria
W fenyloketonurii klasycznej aktywność enzymu, hydroksylazy fenyloalaninowej, w bioptatach komórek wątroby jest niższa niż 1%. W konsekwencji stężenie fenyloalaniny we krwi jest znacznie wyższe. Na zwykłej diecie sięga powyżej poziomu >1200 µmol/l. W tym typie fenyloketonurii pacjenci są niezwykle wrażliwi na wszelkie odstępstwa bądź zaniedbania terapii. Tak wysoki poziom fenyloalaniny przyczynia się do licznych zaburzeń neurologicznych, takich jak zaburzenia mielinizacji i istoty białej układu nerwowego. U nieleczonych niemowląt zauważalne są także zaburzenia w istocie szarej, przede wszystkim w korze ruchowej, przedruchowej, wzgórzu i hipokampie.
Łagodna fenyloketonuria
Do nieco łagodniejszych postaci choroby należy łagodna fenyloketonuria, w której aktywność enzymu hydroksylazy fenyloalaninowej wynosi o 1 do 3%. Dzięki temu stężenie fenyloalaniny we krwi u tych niemowląt na zwykłej diecie jest na poziomie 600-1200 µmol/l. Ci pacjenci wykazują się nieco wyższą tolerancją na wysokie stężenia fenyloalaniny we krwi, jednak nadal do prawidłowego rozwoju niezbędna jest odpowiednia terapia i unikanie fenyloalaniny.
Hiperfenyloalaninemia
Zdecydowanie najłagodniejszym typem fenyloketonurii jest łagodna hiperfenyloalaninemia (HPA). W przypadku HPA aktywność enzymu odpowiedzialnego za metabolizm fenyloalaniny sięga nawet od 3 do 6%. W przypadku pacjentów z łagodną hiperfenyloalaninemią stężenie fenyloalaniny we krwi na zwykłej diecie zazwyczaj nie przekracza 600 µmol/l. Jednakże nadal, do prawidłowego rozwoju dzieci, ważne jest przestrzeganie odpowiednio dobranej terapii i utrzymywanie poziomu fenyloalaniny we krwi na ustalonych wartościach.
Fenyloketonuria matczyna
Nieco innym rodzajem fenyloketonurii na jaką narażone są niemowlęta jest fenyloketonuria matczyna. Występuje ona, gdy na fenyloketonurię choruje kobieta ciężarna i nie przestrzega odpowiedniej diety eliminacyjnej. W tej sytuacji również występuje brak aktywności enzymu hydroksylazy fenyloalaninowej. Jeśli mama nie przestrzega terapii, dochodzi do podniesienia poziomu fenyloalaniny we krwi, co może przyczynić się do uszkodzenia płodu. U takich niemowląt najczęściej obserwuje się przede wszystkim opóźnienie rozwoju układu nerwowego, zaburzenia rozwoju umysłowego i motorycznego, a także wady serca.
Więcej o zespole fenyloketonurii matczynej przeczytasz tutaj.
Fenyloketonuria - częstość występowania
Częstość występowania fenyloketonurii w Polsce, podobnie jak w innych krajach Europy Środkowej, wynosi od 1:7000 do 1:8000. Jest to najczęściej występująca choroba wśród wrodzonych wad metabolizmu. Częstość występowania łagodnych postaci hiperfenyloalaninemii w populacji polskiej jest około 5 razy mniejsza w porównaniu z postacią klasyczną.
Rozpoznawanie i leczenie fenyloketonurii
W latach 30. ubiegłego stulecia Norweg Asbjorn Folling, lekarz i biochemik, oznaczył w moczu chorych z upośledzeniem umysłowym kwas fenylopirogronowy, co przyczyniło się do wykrycia genetycznie uwarunkowanej choroby zwanej wówczas „imbecillitas phenylpyruvica" (nazwę zmieniono później na fenyloketonurię lub PKU).
W 1951 r. niemiecki lekarz Horst Bickel rozpoczął pracę nad wytworzeniem pierwszej bezfenyloalaninowej odżywki dla chorego dziecka. Kolejnym ważnym etapem w badaniach nad fenyloketonurią było znalezienie takiej metody diagnostycznej, by jak najwcześniej móc ją rozpoznawać. To Amerykanin Robert Guthrie, mikrobiolog, w 1957 roku opracował prosty test do mierzenia poziomu fenyloalaniny we krwi na podstawie kilku kropli krwi pobranych u wszystkich noworodków (w 1961 roku test ten został uproszczony). Już wtedy wiadomo było, że im szybciej zostanie wprowadzona dieta niskofenyloalaninowa, tym mniej dotkliwe będą skutki choroby.
Robert Guthrie jest ojcem badania przesiewowego w kierunku rozpoznawania fenyloketonurii. W 1965 roku profesor Barbara Cabalska z Instytutu Matki i Dziecka w Warszawie rozpoczęła badania przesiewowe w kierunku Fenyloketonurii, stosując test Guthriego. Do 1998 roku badaniem wykonywanym w kierunku tej choroby genetycznej był wspomniany test, obecnie jest to badanie kolorymetryczne i określa stężenia fenyloalaniny we krwi. Dzięki tym osiągnięciom u bardzo wielu dzieci rozpoznaje się fenyloketonurię w pierwszych dniach życia, a poprzez wczesne wprowadzenie postępowania dietetycznego ich życie staje normalne i bezobjawowe.
Jak diagnozuje się fenyloketonurię?
PKU rozpoznawana jest w Polsce w obowiązkowych badaniach przesiewowych wszystkich noworodków, w pierwszych dniach po urodzeniu. Diagnoza choroby stawiana jest na podstawie analiz laboratoryjnych: badania krwi i moczu oraz testów genetycznych.
W pierwszym rzucie wykonywany jest test suchej kropli krwi pobierany na specjalną bibułę filtracyjną, który wskazuje podwyższone stężenie fenyloalaniny (Phe), a obniżone tyrozyny (Tyr). Test moczowy z chlorkiem żelaza w okresie noworodkowym jest testem pomocniczym - kwas fenylopirogronowy odpowiedzialny za dodatni wynik badania pojawia się w moczu w ciągu pierwszych tygodni życia, co jest uzależnione od zawartości fenyloalaniny we krwi (> 15 mg%).
Dzięki testom bibułowym stało się możliwe wdrożenie diety niskofenyloalaninowej jeszcze przed wystąpieniem klinicznych objawów choroby i uchronienie dzieci przed nieodwracalnym uszkodzeniem mózgu. Utrzymanie odpowiedniej diety gwarantuje prawidłowy rozwój intelektualny. Według danych amerykańskich każdy tydzień opóźnienia w rozpoczęciu postępowania dietetycznego skutkuje obniżeniem ilorazu inteligencji o jeden punkt.
Jakie są objawy fenyloketonurii?
Objawy choroby są skutkiem gromadzenia się w organizmie nadmiaru fenyloalaniny i jej metabolitów (kwasu fenylopirogronowego i fenylooctowego). Przy dużych stężeniach, fenyloalanina i jej metabolity wykazują działanie toksyczne na ośrodkowy układ nerwowy.
W dzisiejszych czasach leczeni pacjenci z fenyloketonurią już nie muszą być skazani na takie zagrożenia dla ich zdrowia i życia. Los chorych zmienił się, od kiedy wprowadzono wczesną (poporodową) diagnostykę i natychmiastowe leczenie od pierwszych dni życia. Zawdzięczamy to wszystko kilku dociekliwym lekarzom, których osiągnięcia skutkują dziś prawidłowym rozwojem mózgu u pacjentów z PKU. Objawy choroby są związane z nieprawidłowym funkcjonowaniem mózgu i różnorodnymi groźnymi konsekwencjami, szczególnie w zakresie niepełnosprawności intelektualnej. W piśmiennictwie u nieleczonych chorych z fenyloketonurią opisywane są objawy ciężkiej niepełnosprawności intelektualnej (nierzadko z prawie niemożliwymi do oceny wartościami ilorazu inteligencji), z małogłowiem i charakterystycznymi nieprawidłowościami w budowie twarzy (w tym osadzenie oczu i uszu oraz kształt nosa i ust).
Często wymieniane są też nasilone napady padaczkowe, zaburzenia zachowania (nadpobudliwość lub autyzm, napady agresji, samookaleczanie). Do tego wszystkiego dołączają się zaburzenia chodu, a najciężej dotknięci chorzy, szczególnie w późniejszych dekadach życia, poruszają się jedynie z pomocą osób trzecich lub są trwale unieruchomieni w łóżku.
Ciężkie zaburzenia mowy skutkują całkowitym brakiem możliwości porozumiewania się, a postępujące wraz z wiekiem zaburzenia neurologiczne, w tym niedowłady czterokończynowe uniemożliwiają funkcjonowanie w zakresie podstawowych czynności życiowych. Tacy chorzy wymagają całkowitej opieki ze strony osób trzecich i są często pensjonariuszami domów opieki społecznej.
Dodatkowe objawy fenyloketonurii, związane z tzw. zjawiskiem „rozcieńczania” barwnika, pod postacią jasnych włosów i skóry oraz jasnoniebieskich tęczówek oka dopełniają obraz choroby typowy dla nieleczonych chorych z fenyloketonurią. Nie można też zapomnieć o kłopotliwych trądzikowych zmianach skórnych i wyczuwalnym przykrym „mysim” zapachu potu i moczu. Te wszystkie objawy dotyczą chorych z fenyloketonurią, u których dieta PKU nie była stosowana i zwykle choroba była rozpoznawana za późno.
Dlaczego chorujemy? Dziedziczenie fenyloketonurii
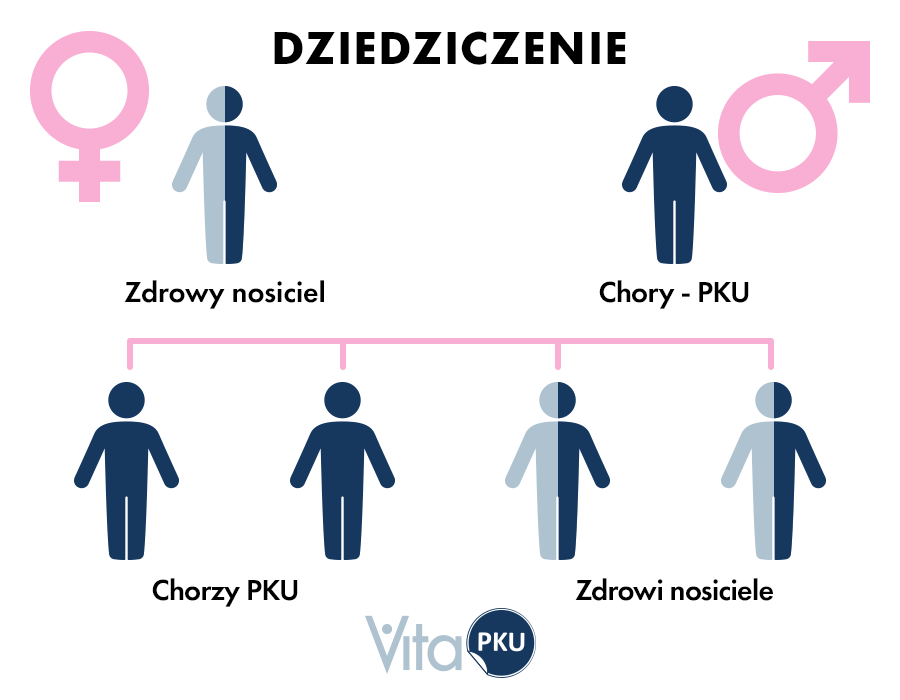
Za rozwój tej choroby odpowiada predyspozycja genetyczna. Fenyloketonuria jest dziedziczona autosomalnie, recesywnie. W praktyce oznacza to, że za rozwój schorzenia odpowiedzialna jest mutacja genu, która występuje u obojga rodziców. Dziedziczenie autosomalne związane jest z chromosomami innymi niż chromosomy płci. Ponadto dziedziczenie recesywne oznacza, że dana mutacja jest przekazana od obydwojga rodziców. Warto podkreślić, że właśnie u rodziców mutacja ta może być niezauważalna. Mogą być oni heterozygotyczni pod względem tej cechy, czyli posiadają tę mutację, ale tylko na jednym chromosomie i są nosicielami. Jeśli dwie takie osoby (nosiciele) zostaną rodzicami i przekażą dziecku chromosom z mutacją, dochodzi do rozwoju fenyloketonurii.
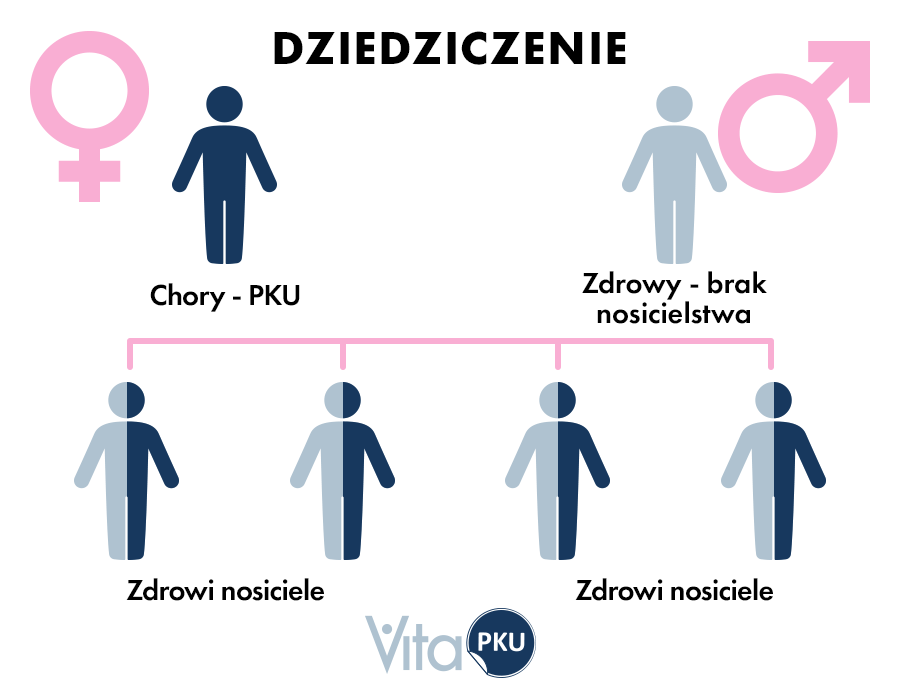
Wspomniana mutacja najczęściej dotyczy enzymów biorących udział w przemianie fenyloalaniny. Powoduje to brak obecności lub brak aktywności tego enzymu. W konsekwencji przyczynia się to do gromadzenia fenyloalaniny w nadmiernych ilościach i uszkodzenia układu nerwowego.
Czym jest fenyloalanina?
Wiemy, że fenyloalanina (Phe) to aminokwas egzogenny (zewnątrzpochodny) i dostarczany jest przez pożywienie jako białko pokarmowe. Jej wysokie stężenia w organizmie prowadzą do uszkodzenia mózgu i dlatego leczenie pacjentów z rozpoznaną fenyloketonurią to ciągła konieczność zmniejszenia ilości fenyloalaniny w ich diecie.
Dieta niskofenyloalaninowa (od najwcześniejszego okresu życia tj. od okresu noworodkowego, aż do końca życia) zapobiega pojawieniu się jednej z najcięższych form niepełnosprawności intelektualnej, jaką jeszcze spotyka się u nieleczonych dorosłych chorych z późno rozpoznaną fenyloketonurią (pacjentów nieobjętych przesiewowymi badaniami noworodkowymi). Udowodniono, że tylko tak można ochronić ośrodkowy układ nerwowy przed nadmiarem fenyloalaniny.
Dlaczego wysokie stężenie fenyloalaniny jest niebezpieczne dla mózgu?
Czy pacjent z PKU, u którego wcześnie rozpoznano chorobą i stosowano dietę, może czuć się bezpiecznie, jeżeli odstępuje od diety niskofenylolaninowej? Czy wtedy jego choroba - fenyloketonuria może jeszcze spowodować niepokojące objawy? Przyjrzyjmy się temu, co dzieje się w mózgu w przypadku wysokich stężeń fenyloalaniny.
Gdy dochodzi do niekontrolowanego nadmiaru fenyloalaniny w diecie i potem we krwi chorego, to aminokwas ten przedostaje się przez barierę krew-mózg do ośrodkowego układu nerwowego (OUN). Doprowadza to wtedy do uruchomienia złożonych mechanizmów, które uszkadzają mózg. Z jednej strony przyczynia się to do obniżenia stężeń innych aminokwasów (ważnych dla rozwoju i funkcjonowania komórek mózgu zwanych neuronami) i produkcji neuroprzekaźników, a z drugiej strony – do zahamowania produkcji mieliny (zewnętrznej otoczki włókna nerwowego w mózgu).
Tyrozyna i tryptofan – dlaczego są ważne dla pacjentów z PKU?
Dla rozwoju i funkcjonowania mózgu istotne znaczenie dla pacjentów z PKU mają też inne aminokwasy, w tym szczególnie tyrozyna i tryptofan. To z ich dalszych przemian powstają neuroprzekaźniki. Są to substancje, które pośredniczą w przekazywaniu informacji między poszczególnymi komórkami mózgu. I tak z tyrozyny powstaje dopamina, a z tryptofanu – serotonina. Nietrudno przewidzieć, że w przypadku ich niedoboru spowodowanego nadmiarem fenyloalaniny będzie dochodzić do nieprawidłowego funkcjonowania mózgu.
Pojawiają się wtedy zaburzenia wynikające z niedoboru dopaminy. Są to: rozkojarzenie, trudności z koncentracją, problemy z zapamiętywaniem i porządkowaniem informacji, senność, zmęczenie w ciągu dnia, depresja, spowolnienie ruchów, drżenie kończyn w stanie spoczynku. Udowodniono, że niektóre obszary mózgu, zwłaszcza okolica kory przedczołowej, są w szczególności wrażliwe na niedobór dopaminy.
Zaobserwowano też, że u źle kontrolowanych i nieleczonych pacjentów z PKU dochodzi do dysfunkcji tego obszaru mózgu i pojawiają się niekorzystne zaburzenia wyższych funkcji nerwowych. Dodatkowo też dochodzi do niedoboru wymienionego wcześniej neuroprzekaźnika, jakim jest serotonina, zwana „hormonem szczęścia”. W przypadku jej deficytu pojawiają się objawy takie jak: długotrwały spadek nastroju, smutek, zmęczenie, skłonność do agresji, brak apetytu lub objadanie się (głównie słodyczami, bo cukier jest niezbędny do produkcji serotoniny), przewlekła bezsenność. Mogą też pojawić się obsesje, lęki i zaburzenia depresyjne, a także może zwiększyć się wrażliwość na ból. Wtedy mówimy o tzw. powikłaniach neuropsychologicznych. Mogą pojawić się w każdym okresie życia chorego z PKU, który nie kontroluje diety ubogofenyloalaninowej albo z niej rezygnuje.
Wpływ nadmiaru fenyloalaniny na tzw. istotę białą mózgu
Nadmiar fenyloalaniny może spowodować też inne nieprawidłowości OUN w tzw. istocie białej mózgu. Dotyczy to zaburzeń mielinizacji włókien nerwowych OUN. Dochodzi wtedy do zahamowania produkcji mieliny przez oligodendrocyty (komórki mózgu) i do nadmiernego jej rozpadu. Taki proces zwany demielinizacją doprowadza ostatecznie do zmniejszenia ilości osłonek mielinowych włókien nerwowych mózgu, ale w pewnym stopniu może być jeszcze odwracalny.
W zależności od intensywności procesów dysmielinizacyjnych różne mogą być następstwa. Wszystko uzależnione jest od tego, jakim zasięgiem tego procesu są objęte neurony (komórki mózgu) oraz jakimi będzie to skutkować zaburzeniami w zakresie przewodnictwa między neuronami i ich połączeniami. Zmiany te stwierdza się u niezbilansowanych dietetycznie chorych z fenyloketonurią i można je uwidocznić w badaniu rezonansu magnetycznego głowy. Takie nieprawidłowości mielinizacji w istocie białej mózgu powodują zaburzenia intelektualne. Są to: trudności w nauce i przyswajaniu informacji, zaburzenia pamięci, a także różnie nasilone deficyty intelektualne.
Neurotoksyczność fenyloalaniny
Mówimy o tzw. neurotoksyczności fenyloalaniny, ponieważ nadmiar fenyloalaniny (Phe) działa bardzo toksycznie na neurony (komórki mózgu) w każdym wieku chorego z PKU. Te wymienione wcześniej powikłania neuropsychologiczne (rozdrażnienie, utrudniony kontakt z innymi, skłonność do agresywnego zachowania, zaburzenia snu), a także zmiany dysmielinizacyjne w istocie białej (zaburzenia zapamiętywania, deficyty intelektualne) zawsze wynikają z wysokich stężeń fenyloalaniny i wskazują na konieczność eliminacji jej w diecie pacjenta. Każdy pacjent, który nie chce kontynuować diety PKU, jest zagrożony pojawieniem się tych okrutnych następstw.
Nie sposób pominąć tu zagadnienia związanego z toksycznym działaniem nadmiaru fenyloalaniny na mózg płodu, którego matka z PKU nie eliminuje jej podaży podczas trwania ciąży. Mówimy wtedy o zespole fenyloketonurii matczynej (MPKU), który oznacza istnienie choroby u matki, a dotyczy uszkodzenia płodu w przebiegu ciąży kobiety z nadmiernymi stężeniami fenyloalaniny we krwi. Przy stężeniach Phe powyżej 6 mg% kobieta z PKU nie powinna zachodzić w ciążę. Mózg płodu jest szczególnie wrażliwy na podwyższone wartości fenyloalaniny, która działa na jego strukturę jak trucizna. Doprowadza to nieodwracalnego uszkodzenia mózgu, zahamowania jego rozwoju i w następstwie małogłowia oraz głębokich deficytów intelektualnych.
Dziecko z zespołem fenyloketonurii matczynej demonstruje też wady rozwojowe innych narządów, w tym ciężkie wady serca, nieprawidłowości w budowie kończyn i twarzy (dysmorfia), a także zahamowanie wzrostu i niedobór masy ciała. Dysmorfia twarzy jest charakterystyczna i obejmuje: obecność zmarszczki nakątnej, opadającą powiekę, gotyckie podniebienie oraz nisko osadzone, duże, zrotowane ku tyłowi uszy ze słabo rozwiniętą małżowiną. Niestety niepełnosprawność intelektualna pozostaje do końca życia, dołączają się zaburzenia mowy i zachowania (nadpobudliwość ruchowa ADHD) oraz pogłębiają się deficyty pamięci.
Dieta dzieci i dorosłych chorujących na fenyloketonurię
U noworodka chorego na fenyloketonurię, w momencie urodzenia poziom fenyloalaniny w płynach ustrojowych jest prawidłowy - odpowiedzialny za to zjawisko jest enzym matki, którego aktywność jest wystarczająca dla wyrównania defektu enzymatycznego płodu. Jednak już w pierwszych dniach po urodzeniu zaburzenia przemiany fenyloalaniny u chorego noworodka powodują szybkie narastanie stężenia tego aminokwasu w płynach ustrojowych. Fenyloalanina jest aminokwasem egzogennym, czyli takim który musi być dostarczany z dietą. Jest konieczny do prawidłowej biosyntezy białek budulcowych organizmu oraz do syntezy związków, których jest prekursorem (aminy biogenne, substancje barwnikowe, hormony tarczycy). Dlatego schemat diety musi uwzględniać podstawowe zapotrzebowanie na ten aminokwas.
Zapotrzebowanie na fenyloalaninę
Zapotrzebowanie na fenyloalaninę zależy przede wszystkim od tolerancji związanej ze stopniem aktywności hydroksylazy fenyloalaninowej i w związku z tym schemat diety musi być w każdym przypadku dostosowany indywidualnie. Dziecko jest regularnie poddawane kontroli stężenia fenyloalaniny we krwi i według norm lekarz lub wyspecjalizowany w tej chorobie dietetyk ustala precyzyjnie, ile fenyloalaniny dziennie może być w diecie w danym okresie, aż do następnego badania. Szczególnie ważne jest stosowanie ścisłej diety do momentu zakończenia mielinizacji układu nerwowego tj. do ok. 10. roku życia, pamiętając, że postępowanie dietetyczne trwa całe życie, ale z czasem może być nieco mniej restrykcyjne.
Preparaty – podstawa diety dzieci z fenyloketonurią
Żywienie dzieci z fenyloketonurią bazuje na stosowaniu preparatów nisko- lub bezfenyloalaninowych, produkowanych na bazie hydrolizatów białkowych lub pozbawione fenyloalaniny syntetyczne mieszaniny aminokwasów uzupełniane w witaminy, składniki mineralne, pierwiastki śladowe. W ostatnich latach rynek takich preparatów uległ bardzo dużemu wzbogaceniu. Należy pamiętać jednak, że preparaty nie mogą stanowić jedynego źródła pożywienia dziecka, ani osoby dorosłej. Z preparatem PKU trzeba łączyć inne dozwolone produkty niskobiałkowe stosownie do wieku, aby dieta była pełnowartościowa. Bardzo ważne jest, żeby preparat był spożywany codziennie, w dawce zapisanej przez lekarza lub dietetyka. Preparat i dieta są „lekarstwem” na chorobę. Z czasem preparat zmienia się na bardziej skoncentrowany, dostosowany do wieku osoby chorej na PKU. Wybór postaci (proszek, płyn) i smaków jest spory, więc każdy może wybrać dla siebie najlepszy.
Dieta niskobiałkowa i jej zasady
Tak jak wspomniano wyżej, w fenyloketonurii stosowana dieta jest podstawowym narzędziem terapeutycznym choroby. Odpowiednio skomponowany jadłospis umożliwia dzieciom prawidłowy wzrost i rozwój, a także funkcjonowanie zbliżonego do normalnego. Jest to szczególnie ważne w przypadku niemowląt i małych dzieci, u których to układ nerwowy dopiero się kształtuje.
W fenyloketonurii stosuje się dietę niskofenyloalaninową, aby ograniczyć negatywny wpływ aminokwasu na układ nerwowy. Ten model żywienia obowiązuje chorych przez całe życie. Podaż fenyloalaniny w codziennej diecie jest ustalana dla każdego pacjenta indywidualnie i zmienia się wraz z wiekiem. Warto podkreślić, że poziom fenyloalaniny w diecie nie może być ani zbyt wysoki, ani także zbyt niski. Zbyt wysoki, tak jak już wyżej wspomniano, prowadzi do uszkodzenia ośrodkowego układu nerwowego, a zbyt niski zaburza prawidłowy rozwój dziecka np. zaburza funkcjonowanie tarczycy czy układu mięśniowego.
W tym modelu żywienia należy unikać produktów, które są najbardziej zasobne w fenyloalaninę. Są to artykuły takie jak: mięso, ryby czy nabiał (mleko i przetwory mleczne). Właśnie te produkty należy zastępować w diecie specjalnymi preparatami PKU, dedykowanymi osobom z fenyloketonurią. Produkty PKU zawierają znacznie mniej fenyloalaniny lub nie zawierają jej wcale. Warto także podkreślić, że dzieci na diecie niskofenyloalaninowej powinny mieć nieco wyższą podaż białka (hydrolizaty białkowe są gorzej wykorzystywane) i energii. Listę produktów dozwolonych i zabronionych znajdziesz poniżej:
|
PODZIAŁ PRODUKTÓW SPOŻYWCZYCH W DIECIE NISKOFENYLOALANINOWEJ |
|
|
DOZWOLONE |
ZABRONIONE |
|
|
Jak łatwo można zauważyć, dieta niskofenyloalaninowa to również dieta niskobiałkowa. Z tego względu przygotowanie pełnowartościowego i smacznego posiłku może być niezwykle trudne. W końcu białko to podstawowy budulec dla układu odpornościowego czy tkanki mięśniowej. Zatem bardzo ważne jest umiejętne skomponowanie diety. Warto poradzić się doświadczonego dietetyka jak prawidłowo komponować jadłospis niemowlęcia, a później także starszego dziecka.
Należy także podkreślić, że codzienna dieta powinna być na tyle zróżnicowana, na ile jest to możliwe. Trzeba korzystać z różnych owoców, warzyw czy produktów niskobiałkowych takich jak ryż czy ziemniaki. Do tego niezbędne jest włączenie preparatów dedykowanych fenyloketonurii, produktów PKU. Ich bardzo ważną rolą jest dostarczenie pozostałych aminokwasów, energii, witamin i składników mineralnych, a ograniczenie podaży fenyloalaniny. Obecnie asortyment żywności PKU znacznie się powiększył i dostępne są już takie produkty jak chleb niskobiałkowy, niskobiałkowa mąka czy makaron, a także zastępniki: jajek, mięsa czy ryb.
Warto podkreślić, że mimo, iż restrykcje dietetyczne obowiązują całe życie, to spożywanie produktów dozwolonych, zamienników PKU i specjalnych preparatów PKU pozwala na skomponowanie prawidłowej i dobrze zbilansowanej diety, a w konsekwencji także prawidłowy wzrost, rozwój i funkcjonowanie.
Zobacz także: 6 pomysłów, jak uniknąć dietetycznej monotonii w diecie pku
Czy kobieta z fenyloketonurią może urodzić zdrowe dziecko?
Jeżeli kobieta z PKU stosuje podczas ciąży restrykcyjną dietę niskofenyloalaninową i stężenia fenyloalaniny nie przekraczają maksymalnych wartości 6 mg% (najlepiej 2-4 mg%), to ma szansę urodzić zdrowe dziecko. Wiele kobiet z PKU planuje swoje ciąże, wówczas stosują dietę PKU przynajmniej 3 miesiące wcześniej. Wspaniałą nagrodą jest urodzenie zdrowego dziecka, którego rozwój intelektualny i fizyczny nie odbiega od rozwoju rówieśników.
Wysokie stężenia fenyloalaniny u pacjentów z PKU mogą zawsze przyczyniać się do powstania powikłań w każdym okresie ich życia. Dlatego każdy chory z fenyloketonurią musi pamiętać o swojej chorobie i o tym, że jego mózg jest wrażliwy na nadmiar fenyloalaniny przez całe życie. Znajomość diety niskofenyloalaninowej i jej stosowanie u chorych z PKU oraz stałe pomiary stężeń fenyloalaniny są obecnie tzw. „złotym” środkiem zapobiegającym niebezpiecznym stanom nadmiaru fenyloalaniny i jej toksycznym działaniem na struktury mózgu.
Czytaj więcej: Dieta przyszłej mamy z PKU
Czy niskie stężania fenyloalaniny u chorych z PKU stanowią także zagrożenia dla ich zdrowia?
Zbyt niskie stężenia fenyloalaniny u chorych z PKU to rzadkość. Jednak pacjenci, którzy nie spożywają preparatów aminokwasowych pozbawionych fenyloalaniny i ograniczają ilość produktów białkowych niskofenyloalaninowych, są narażeni na taki stan. Wtedy dochodzi do spadku stężenia białka (z groźnymi konsekwencjami dla organizmu) oraz do powikłań związanych z niedoborem fenyloalaniny.
W prawidłowych warunkach fenyloalanina Phe w organizmie człowieka jest prekursorem hormonów tarczycy, amin biogennych (noradrenaliny i adrenalin), substancji barwnikowych (melanin) i składnikiem białek strukturalnych. Utrzymujące się przez dłuższy okres zbyt małe stężenie fenyloalaniny we krwi może zatem powodować utratę masy ciała, brak barwników (jasne włosy i jasna karnacja) oraz nadpobudliwość, drżenia mięśniowe i wzmożoną gotowość do drgawek.
Ilość białka i fenyloalaniny w diecie musi być indywidualnie dobrana do wieku, płci i masy ciała oraz stanu zdrowia, a także do indywidualnej dobowej tolerancji fenyloalaniny. Dieta ubogofenyloalaninowa była, jest i na pewno na długo pozostanie najlepszą formą terapii PKU. Okazuje się, że najlepsze efekty w stosowaniu diety mają pacjenci, którzy biorą udział w szkoleniach, programach edukacyjnych i wspierają się nawzajem.
Nie zmarnujmy szansy, jaką przyniosły badania przesiewowe noworodków w kierunku wczesnego wykrywania fenyloketonurii, jak i możliwość włączenia skutecznej diety niskofenyloalaninowej – jeszcze przed pojawieniem się objawów tej choroby! Każdy pacjent, jeśli tylko stosuje dietę PKU, ma takie same szanse na rozwój intelektualny i karierę jak jego zdrowi koledzy. Niebezpieczne dla mózgu są zarówno zbyt wysokie, jak i zbyt niskie stężenia Phe.
Pamiętajmy, że ta wrażliwość mózgu na stężenia Phe utrzymuje się u chorego z PKU przez całe życie. Systematyczne postępowanie dietetyczne w PKU jest ukoronowane sukcesem: prawidłowym rozwojem i czynnością mózgu do końca życia. Świadomość pacjenta z PKU o istocie i zagrożeniach choroby oraz o konieczności stosowania kontrolowanego leczenia dietetycznego przez całe życie pozwala pokonać wszystkie przeszkody, by uchronić mózg od nadmiaru fenyloalaniny i móc być szczęśliwym, spełniać swoje marzenia.
Bibliografia:
- Choroby i wady wykrywane w badaniach przesiewowych, Instytut Matki i Dziecka, Zakład Badań przesiewowych, http://przesiew.imid.med.pl/fenyloketonuria.html
- Fenyloketonuria – diagnostyka, leczenie, wybrane problemy kliniczne, lek. Bożena Didycz, dr hab. n. med. Mirosław Bik-Multanowski, prof. UJ, Pediatria po Dyplomie, 2017, 02.
- Centrum Badań Genetycznych, www.genesis.pl Narodowe Centrum Edukacji Żywieniowej, Instytut Żywności i Żywienia, https://ncez.pl/dzieci-i-mlodziez/dzieci-0-3/fenyloketonuria-----rzadka-choc-czesta-wada-metabolizmu
- Podstawy naukowe żywienia w szpitalach. M. Jarosz i wsp. Instytut Żywności i Żywienia, 2001.
- Żywienie człowieka zdrowego i chorego cz. 2. M. Grzmisławski i wsp. Wydawnictwo Naukowe PWN 2012.



